metan v1.10.0 now on CRAN
I’m very pleased to announce that metan 1.10.0 is now on
CRAN. Some minor improvements and new functions were added in this version.
Instalation
# The latest stable version is installed with
install.packages("metan")
# Or the development version from GitHub:
# install.packages("devtools")
devtools::install_github("TiagoOlivoto/metan")
New functions
get_dist()to get distance matrices from objects of classclustering.
library(metan)
# Registered S3 method overwritten by 'GGally':
# method from
# +.gg ggplot2
# |========================================================|
# | Multi-Environment Trial Analysis (metan) v1.11.0 |
# | Author: Tiago Olivoto |
# | Type 'citation('metan')' to know how to cite metan |
# | Type 'vignette('metan_start')' for a short tutorial |
# | Visit 'https://bit.ly/2TIq6JE' for a complete tutorial |
# |========================================================|
d <- data_ge2 %>%
means_by(GEN) %>%
column_to_rownames("GEN") %>%
clustering()
get_dist(d)$d %>% round_cols(digits = 1)
# H1 H10 H11 H12 H13 H2 H3 H4 H5 H6 H7 H8 H9
# H1 0.0 49.2 36.6 55.9 39.8 22.0 28.6 34.5 42.5 10.8 25.0 50.6 63.5
# H10 49.2 0.0 13.7 8.3 43.0 48.9 29.2 46.0 48.1 51.3 26.9 8.8 16.6
# H11 36.6 13.7 0.0 20.0 40.1 40.2 16.2 41.0 45.3 40.2 13.6 14.4 27.1
# H12 55.9 8.3 20.0 0.0 49.1 56.0 34.0 52.9 54.4 58.5 32.7 8.6 9.7
# H13 39.8 43.0 40.1 49.1 0.0 20.8 48.0 9.1 6.8 32.5 40.8 49.9 58.2
# H2 22.0 48.9 40.2 56.0 20.8 0.0 40.9 14.2 22.0 12.7 34.5 53.6 64.9
# H3 28.6 29.2 16.2 34.0 48.0 40.9 0.0 46.8 53.0 36.1 7.9 26.5 39.3
# H4 34.5 46.0 41.0 52.9 9.1 14.2 46.8 0.0 8.7 26.2 39.7 52.5 61.9
# H5 42.5 48.1 45.3 54.4 6.8 22.0 53.0 8.7 0.0 34.1 45.6 55.2 63.3
# H6 10.8 51.3 40.2 58.5 32.5 12.7 36.1 26.2 34.1 0.0 30.9 54.4 66.7
# H7 25.0 26.9 13.6 32.7 40.8 34.5 7.9 39.7 45.6 30.9 0.0 26.2 39.2
# H8 50.6 8.8 14.4 8.6 49.9 53.6 26.5 52.5 55.2 54.4 26.2 0.0 13.2
# H9 63.5 16.6 27.1 9.7 58.2 64.9 39.3 61.9 63.3 66.7 39.2 13.2 0.0
get_corvars()to get normal, multivariate correlated variables.
sigma <- matrix(c(1, .3, 0,
.3, 1, .9,
0, .9, 1),3,3)
mu <- c(6,50,5)
df <- get_corvars(n = 10000, mu = mu, sigma = sigma, seed = 101010)
means_by(df)
# # A tibble: 1 x 3
# X1 X2 X3
# <dbl> <dbl> <dbl>
# 1 6.01 50.0 5.00
cor(df)
# X1 X2 X3
# X1 1.000000000 0.3026515 0.004851063
# X2 0.302651488 1.0000000 0.900604867
# X3 0.004851063 0.9006049 1.000000000
get_covmat()to obtain covariance matrix based on variances and correlation values.
cormat <-
matrix(c(1, 0.9, -0.4,
0.9, 1, 0.6,
-0.4, 0.6, 1),
nrow = 3,
ncol = 3)
get_covmat(cormat, var = c(16, 25, 9))
# V1 V2 V3
# V1 16.0 18 -4.8
# V2 18.0 25 9.0
# V3 -4.8 9 9.0
as_numeric(),as_integer(),as_logical(),as_character(), andas_factor()to coerce variables to specific formats quickly.
library(metan)
library(tibble)
# Warning: package 'tibble' was built under R version 4.0.3
df <-
tibble(y = rnorm(5),
x1 = c(1:5),
x2 = c(TRUE, TRUE, FALSE, FALSE, FALSE),
x3 = letters[1:5],
x4 = as.factor(x3))
df
# # A tibble: 5 x 5
# y x1 x2 x3 x4
# <dbl> <int> <lgl> <chr> <fct>
# 1 -1.59 1 TRUE a a
# 2 -1.62 2 TRUE b b
# 3 -0.751 3 FALSE c c
# 4 1.13 4 FALSE d d
# 5 0.472 5 FALSE e e
# Convert y to integer
as_integer(df, y)
# # A tibble: 5 x 5
# y x1 x2 x3 x4
# <int> <int> <lgl> <chr> <fct>
# 1 -1 1 TRUE a a
# 2 -1 2 TRUE b b
# 3 0 3 FALSE c c
# 4 1 4 FALSE d d
# 5 0 5 FALSE e e
# convert x3 to factor
as_factor(df, x3)
# # A tibble: 5 x 5
# y x1 x2 x3 x4
# <dbl> <int> <lgl> <fct> <fct>
# 1 -1.59 1 TRUE a a
# 2 -1.62 2 TRUE b b
# 3 -0.751 3 FALSE c c
# 4 1.13 4 FALSE d d
# 5 0.472 5 FALSE e e
# Convert all columns to character
as_character(df, everything())
# # A tibble: 5 x 5
# y x1 x2 x3 x4
# <chr> <chr> <chr> <chr> <chr>
# 1 -1.59386583725876 1 TRUE a a
# 2 -1.61771631653143 2 TRUE b b
# 3 -0.751337714367338 3 FALSE c c
# 4 1.13100276654771 4 FALSE d d
# 5 0.472487882906806 5 FALSE e e
# Convert x2 to numeric and coerce to a vector
as_numeric(df, x2, .keep = "used", .pull = TRUE)
# [1] 1 1 0 0 0
n_valid(),n_missing(), andn_unique()to count valid, missing, and unique values, respectively.
library(metan)
data_naz <- iris %>%
group_by(Species) %>%
doo(~head(., n = 3)) %>%
as_character(Species)
data_naz
# # A tibble: 9 x 5
# Species Sepal.Length Sepal.Width Petal.Length Petal.Width
# <chr> <dbl> <dbl> <dbl> <dbl>
# 1 setosa 5.1 3.5 1.4 0.2
# 2 setosa 4.9 3 1.4 0.2
# 3 setosa 4.7 3.2 1.3 0.2
# 4 versicolor 7 3.2 4.7 1.4
# 5 versicolor 6.4 3.2 4.5 1.5
# 6 versicolor 6.9 3.1 4.9 1.5
# 7 virginica 6.3 3.3 6 2.5
# 8 virginica 5.8 2.7 5.1 1.9
# 9 virginica 7.1 3 5.9 2.1
data_naz[c(2:3, 6, 8), c(1:2, 4, 5)] <- NA
n_valid(data_naz)
# Warning: NA values removed to compute the function. Use 'na.rm = TRUE' to
# suppress this warning.
# Warning: NA values removed to compute the function. Use 'na.rm = TRUE' to
# suppress this warning.
# Warning: NA values removed to compute the function. Use 'na.rm = TRUE' to
# suppress this warning.
# # A tibble: 1 x 4
# Sepal.Length Sepal.Width Petal.Length Petal.Width
# <int> <int> <int> <int>
# 1 5 9 5 5
n_missing(data_naz, na.rm = TRUE) # na.rm = TRUE to suppress the warning.
# # A tibble: 1 x 5
# Species Sepal.Length Sepal.Width Petal.Length Petal.Width
# <int> <int> <int> <int> <int>
# 1 4 4 0 4 4
n_unique(data_naz, na.rm = TRUE) # na.rm = TRUE to suppress the warning.
# # A tibble: 1 x 5
# Species Sepal.Length Sepal.Width Petal.Length Petal.Width
# <int> <int> <int> <int> <int>
# 1 4 6 6 6 6
env_stratification()to perform environment stratification using factor analysis.
model <-
env_stratification(data_ge,
env = ENV,
gen = GEN,
resp = everything())
gmd(model)
# Class of the model: env_stratification
# Variable extracted: env_strat
# # A tibble: 28 x 7
# TRAIT ENV MEGA_ENV MEAN MIN MAX CV
# <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
# 1 GY E1 ME1 2.52 1.97 2.90 13.3
# 2 GY E10 ME1 2.18 1.54 2.57 14.4
# 3 GY E11 ME1 1.37 0.899 1.68 16.4
# 4 GY E12 ME1 1.61 1.02 2 20.3
# 5 GY E13 ME1 2.91 1.83 3.52 16.8
# 6 GY E5 ME1 3.91 3.37 4.81 10.7
# 7 GY E3 ME2 4.06 3.43 4.57 8.22
# 8 GY E6 ME2 2.66 2.34 2.98 7.95
# 9 GY E14 ME3 1.78 1.43 2.06 11.7
# 10 GY E8 ME3 2.54 2.05 2.88 10.5
# # ... with 18 more rows
gmd(model, "FA")
# Class of the model: env_stratification
# Variable extracted: FA
# # A tibble: 28 x 9
# TRAIT ENV FA1 FA2 FA3 FA4 FA5 COMMUNALITY UNIQUENESSES
# <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 GY E1 -0.881 0.327 0.00927 -0.0631 0.274 0.963 0.0369
# 2 GY E10 -0.942 -0.158 -0.0820 0.113 0.174 0.962 0.0380
# 3 GY E11 -0.929 -0.233 -0.0336 -0.242 0.110 0.989 0.0111
# 4 GY E12 -0.848 0.135 0.0263 0.0941 0.241 0.805 0.195
# 5 GY E13 -0.940 0.108 -0.0842 -0.0637 -0.235 0.961 0.0391
# 6 GY E14 -0.150 -0.123 -0.916 -0.0872 0.265 0.954 0.0463
# 7 GY E2 -0.198 -0.0521 -0.126 -0.969 0.0328 0.997 0.00266
# 8 GY E3 -0.0806 0.910 0.341 -0.0173 -0.110 0.963 0.0370
# 9 GY E4 0.209 0.543 -0.272 -0.728 -0.120 0.957 0.0433
# 10 GY E5 -0.777 0.392 -0.269 -0.0470 -0.267 0.904 0.0963
# # ... with 18 more rows
Minor improvements
gamem(),gamem_met(), andwaasb()now have abyargument and understand data passed fromgroup_by. Let’s to compute a mixed model within each environment of datasetdata_ge2and extract the variance components for each model.
model <- gamem(data_ge2,
gen = GEN,
rep = REP,
resp = NR:TKW,
by = ENV,
verbose = FALSE)
gmd(model, "vcomp", verbose = FALSE)
# # A tibble: 8 x 7
# ENV Group NR NKR CDED PERK TKW
# <fct> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 A1 GEN 1.14 2.37 0.000821 2.79 513.
# 2 A1 Residual 2.92 6.36 0.000553 2.30 1015.
# 3 A2 GEN 1.14 5.33 0.000781 3.92 2941.
# 4 A2 Residual 0.941 5.86 0.000414 0.575 674.
# 5 A3 GEN 1.18 2.15 0.000956 3.14 841.
# 6 A3 Residual 1.27 7.80 0.000376 0.867 1018.
# 7 A4 GEN 0.375 2.75 0.000133 0.486 291.
# 8 A4 Residual 1.44 11.4 0.000398 0.896 967.
mtsi()andmgidi()now returns the ranks for the contribution of each factor and understand models fitted withgamem()andwaasb()using thebyargument.
mgidi_mod <- mgidi(model,
mineval = .1, # force a greater number of factors
SI = 40,
verbose = FALSE)
gmd(mgidi_mod, verbose = FALSE)
# # A tibble: 20 x 12
# ENV VAR Factor Xo Xs SD SDperc h2 SG SGperc
# <fct> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 A1 PERK FA1 87.4 87.3 -0.157 -0.179 0.784 -0.123 -0.140
# 2 A1 NKR FA2 33.9 34.4 0.507 1.50 0.528 0.268 0.791
# 3 A1 TKW FA3 360. 365. 4.20 1.17 0.603 2.53 0.703
# 4 A1 NR FA4 16.9 16.9 -0.0210 -0.124 0.539 -0.0113 -0.0670
# 5 A1 CDED FA5 0.576 0.583 0.00655 1.14 0.817 0.00535 0.928
# 6 A2 CDED FA1 0.584 0.586 0.00201 0.345 0.850 0.00171 0.293
# 7 A2 PERK FA1 87.6 88.7 1.02 1.16 0.953 0.971 1.11
# 8 A2 NKR FA2 32.3 32.2 -0.0503 -0.156 0.732 -0.0368 -0.114
# 9 A2 NR FA3 15.8 15.6 -0.153 -0.967 0.784 -0.120 -0.758
# 10 A2 TKW FA4 334. 331. -2.66 -0.798 0.929 -2.47 -0.741
# 11 A3 CDED FA1 0.595 0.588 -0.00680 -1.14 0.884 -0.00601 -1.01
# 12 A3 PERK FA1 87.6 87.1 -0.577 -0.659 0.916 -0.529 -0.603
# 13 A3 NR FA2 15.8 16.5 0.708 4.49 0.736 0.521 3.30
# 14 A3 NKR FA3 30.4 30.7 0.253 0.833 0.452 0.115 0.377
# 15 A3 TKW FA4 318. 325. 7.41 2.33 0.712 5.28 1.66
# 16 A4 TKW FA1 343. 347. 4.46 1.30 0.475 2.12 0.618
# 17 A4 PERK FA2 87.0 87.3 0.265 0.305 0.619 0.164 0.189
# 18 A4 NR FA3 16.0 16.1 0.120 0.746 0.438 0.0524 0.327
# 19 A4 NKR FA4 32.5 32.9 0.483 1.49 0.420 0.203 0.625
# 20 A4 CDED FA5 0.589 0.593 0.00345 0.585 0.500 0.00172 0.293
# # ... with 2 more variables: sense <chr>, goal <dbl>
plot.mtsi()andplot.mgidi()now returns a radar plot by default when usingtype = "contribution".
p1 <- plot(mgidi_mod$data[[1]], type = "contribution")
p2 <- plot(mgidi_mod$data[[1]], type = "contribution", radar = FALSE)
arrange_ggplot(p1, p2)
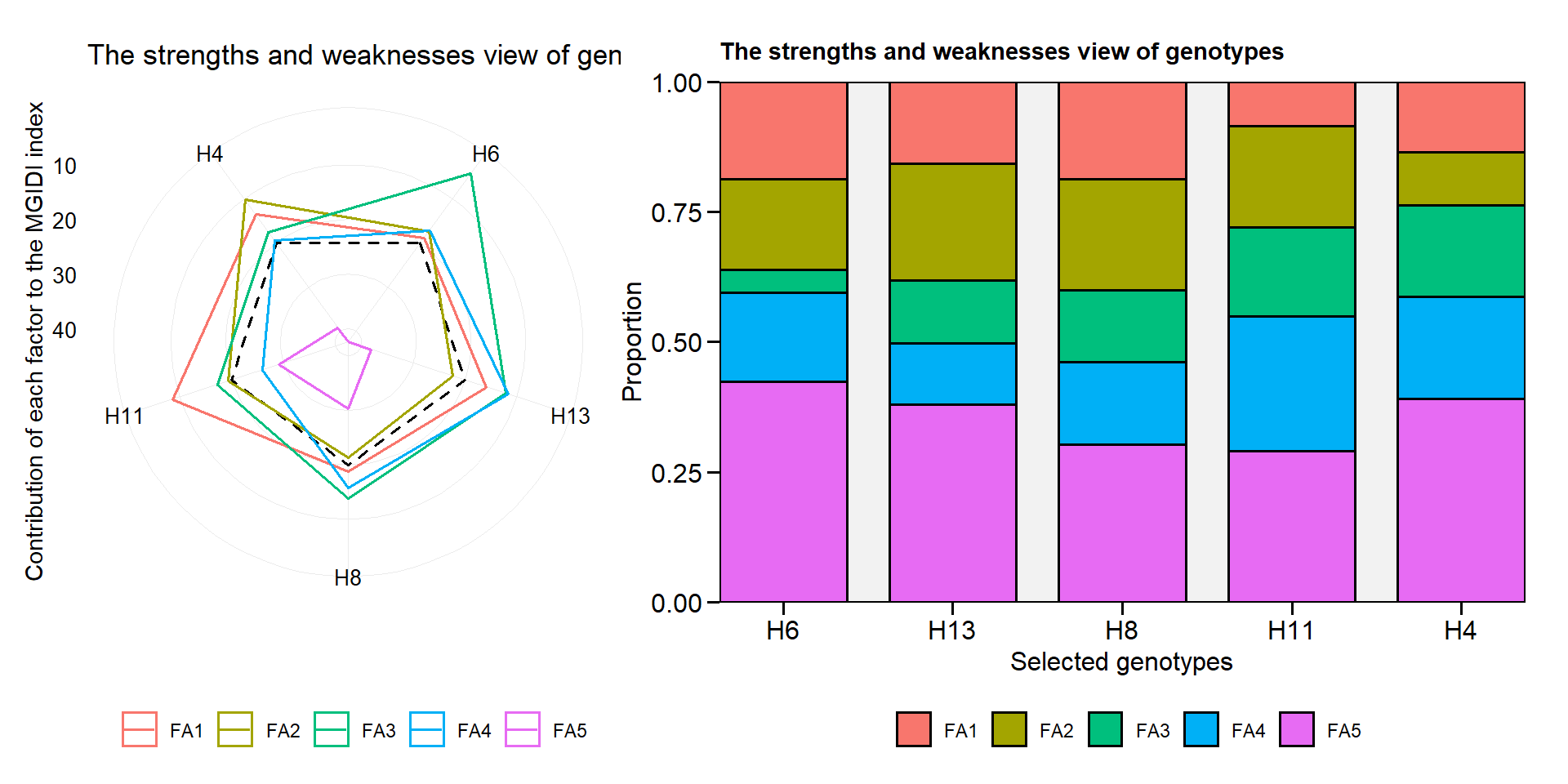
get_model_data()now returns the genotypic and phenotypic correlation matrices from objects of classwaasbandgamem.
(pcor <- gmd(model$data[[1]], "pcor"))
# Class of the model: gamem
# Variable extracted: pcor
# NR NKR CDED PERK TKW
# NR 1.0000000 -0.351700402 -0.3271058 0.344913775 -0.4723885
# NKR -0.3517004 1.000000000 -0.4447736 -0.008091937 -0.1152699
# CDED -0.3271058 -0.444773632 1.0000000 -0.700728407 0.4242704
# PERK 0.3449138 -0.008091937 -0.7007284 1.000000000 -0.2854948
# TKW -0.4723885 -0.115269899 0.4242704 -0.285494825 1.0000000
(gcor <- gmd(model$data[[1]], "gcor"))
# Class of the model: gamem
# Variable extracted: gcor
# NR NKR CDED PERK TKW
# NR 1.0000000 -0.62348035 -0.4359572 0.40578424 -0.37885398
# NKR -0.6234804 1.00000000 -0.4731984 -0.06763518 -0.07922932
# CDED -0.4359572 -0.47319840 1.0000000 -0.80549808 0.51851366
# PERK 0.4057842 -0.06763518 -0.8054981 1.00000000 -0.32151455
# TKW -0.3788540 -0.07922932 0.5185137 -0.32151455 1.00000000
- The new function
make_lower_upper()makes it easier to combine two symmetric matrices into one lower-upper diagonal matrix. In the following example, we’ll put the phenotypic correlation matrix in the lower diagonal and the genotypic correlations in the upper diagonal.
make_lower_upper(pcor, gcor, diag = 1)
# NR NKR CDED PERK TKW
# NR 1.0000000 -0.623480350 -0.4359572 0.40578424 -0.37885398
# NKR -0.3517004 1.000000000 -0.4731984 -0.06763518 -0.07922932
# CDED -0.3271058 -0.444773632 1.0000000 -0.80549808 0.51851366
# PERK 0.3449138 -0.008091937 -0.7007284 1.00000000 -0.32151455
# TKW -0.4723885 -0.115269899 0.4242704 -0.28549482 1.00000000