metan v1.6.0 on CRAN
I’m so excited to announce that the latest version of the R package
metan is now on
CRAN. This is a minor release (v1.6.0) that includes new features and some minor improvements.
New functions
Smith_Hazel()andprint.sh()andplot.sh()for computing the Smith and Hazel selection index.
The Smith-Hazel index is computed with the function Smith_Hazel(). Users can compute the index either by declaring known genetic and phenotypic variance-covariance matrices or by using as inpute data, a model fitted with the function gamem(). In this case, the variance-covariance are extracted internally. The economic weights in the argument weights are set by default to 1 for all traits.
library(metan)
# Registered S3 method overwritten by 'GGally':
# method from
# +.gg ggplot2
# []=====================================================[]
# [] Multi-Environment Trial Analysis (metan) v1.6.0 []
# [] Author: Tiago Olivoto []
# [] Type citation('metan') to know how to cite metan []
# [] Type vignette('metan_start') for a short tutorial []
# [] Visit http://bit.ly/2TIq6JE for a complete tutorial []
# []=====================================================[]
mod <- gamem(data_g, GEN, REP, starts_with("N"))
# Method: REML/BLUP
# Random effects: GEN
# Fixed effects: REP
# Denominador DF: Satterthwaite's method
# ---------------------------------------------------------------------------
# P-values for Likelihood Ratio Test of the analyzed traits
# ---------------------------------------------------------------------------
# model NR NKR NKE
# Complete NA NA NA
# Genotype 0.0056 0.216 0.00952
# ---------------------------------------------------------------------------
# Variables with nonsignificant Genotype effect
# NKR
# ---------------------------------------------------------------------------
smith <- Smith_Hazel(mod)
print(smith)
#
# -----------------------------------------------------------------------------------
# Index coefficients
# -----------------------------------------------------------------------------------
# # A tibble: 3 x 3
# VAR b gen_weights
# <chr> <dbl> <dbl>
# 1 NR -11.84 1
# 2 NKR -9.435 1
# 3 NKE 1.184 1
#
# -----------------------------------------------------------------------------------
# Genetic worth
# -----------------------------------------------------------------------------------
# GEN V1
# 1 H13 142.80619
# 2 H12 120.77661
# 3 H5 115.19274
# 4 H10 89.29844
# 5 H4 88.90470
# 6 H2 86.49310
# 7 H11 84.25171
# 8 H7 62.30464
# 9 H1 51.99961
# 10 H3 51.26071
# 11 H6 51.16514
# 12 H9 50.52352
# 13 H8 48.31407
#
# -----------------------------------------------------------------------------------
# Selection gain
# -----------------------------------------------------------------------------------
# # A tibble: 3 x 8
# VAR Xo Xs SD SDperc h2 SG SGperc
# <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 NR 15.78 16.97 1.189 7.532 0.7359 0.8749 5.543
# 2 NKR 30.40 30.60 0.1960 0.6448 0.4523 0.08866 0.2916
# 3 NKE 467.9 524.9 56.98 12.18 0.7126 40.61 8.678
#
# -----------------------------------------------------------------------------------
# Phenotypic variance-covariance matrix
# -----------------------------------------------------------------------------------
# NR NKR NKE
# NR 1.60 0.13 49
# NKR 0.13 4.75 70
# NKE 48.84 70.12 2782
#
# -----------------------------------------------------------------------------------
# Genotypic variance-covariance matrix
# -----------------------------------------------------------------------------------
# NR NKR NKE
# NR 1.18 -0.18 37
# NKR -0.18 2.15 35
# NKE 36.60 34.72 1982
Minor improvements
get_model_data()now extracts the genotypic and phenotypic variance-covariance matrix from objects of classgamemandwaasb.
get_model_data(mod, "gcov")
# Class of the model: gamem
# Variable extracted: gcov
# NKE NKR NR
# NKE 1982.32081 34.7246852 36.5976336
# NKR 34.72469 2.1466667 -0.1839316
# NR 36.59763 -0.1839316 1.1808547
get_model_data(mod, "pcov")
# Class of the model: gamem
# Variable extracted: pcov
# NR NKR NKE
# NR 1.6045584 0.1303704 48.83903
# NKR 0.1303704 4.7459259 70.11889
# NKE 48.8390313 70.1188889 2781.91453
fai_blup()now returns the total genetic gain and the list with the ideotypes’ construction.
fai <- fai_blup(mod)
#
# -----------------------------------------------------------------------------------
# Principal Component Analysis
# -----------------------------------------------------------------------------------
# eigen.values cumulative.var
# PC1 1.97579911 65.85997
# PC2 0.95352008 97.64397
# PC3 0.07068081 100.00000
#
# -----------------------------------------------------------------------------------
# Factor Analysis
# -----------------------------------------------------------------------------------
# FA1 comunalits
# NR -0.7669378 0.5881937
# NKR -0.6510612 0.4238807
# NKE -0.9816949 0.9637248
#
# -----------------------------------------------------------------------------------
# Comunalit Mean: 0.6585997
# Selection differential
# -----------------------------------------------------------------------------------
# # A tibble: 3 x 9
# VAR Factor Xo Xs SD SDperc h2 SG SGperc
# <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 NR 1 15.8 17.4 1.63 10.3 0.736 1.20 7.60
# 2 NKR 1 30.4 31.2 0.814 2.68 0.452 0.368 1.21
# 3 NKE 1 468. 532. 64.0 13.7 0.713 45.6 9.74
#
# -----------------------------------------------------------------------------------
# Selected genotypes
# H13 H5
# -----------------------------------------------------------------------------------
mgidi()now computes the genetic gain when a mixed-model is used as input data.
mgidi_index <- mgidi(mod)
#
# -------------------------------------------------------------------------------
# Principal Component Analysis
# -------------------------------------------------------------------------------
# # A tibble: 3 x 4
# PC Eigenvalues `Variance (%)` `Cum. variance (%)`
# <chr> <dbl> <dbl> <dbl>
# 1 PC1 1.98 65.9 65.9
# 2 PC2 0.954 31.8 97.6
# 3 PC3 0.0707 2.36 100
# -------------------------------------------------------------------------------
# Factor Analysis - factorial loadings after rotation-
# -------------------------------------------------------------------------------
# # A tibble: 3 x 4
# VAR FA1 Communality Uniquenesses
# <chr> <dbl> <dbl> <dbl>
# 1 NR -0.767 0.588 0.412
# 2 NKR -0.651 0.424 0.576
# 3 NKE -0.982 0.964 0.0363
# -------------------------------------------------------------------------------
# Comunalit Mean: 0.6585997
# -------------------------------------------------------------------------------
# Selection differential
# -------------------------------------------------------------------------------
# # A tibble: 3 x 10
# VAR Factor Xo Xs SD SDperc h2 SG SGperc sense
# <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
# 1 NR FA 1 15.8 17.4 1.63 10.3 0.736 1.20 7.60 increase
# 2 NKR FA 1 30.4 31.2 0.814 2.68 0.452 0.368 1.21 increase
# 3 NKE FA 1 468. 532. 64.0 13.7 0.713 45.6 9.74 increase
# ------------------------------------------------------------------------------
# Selected genotypes
# -------------------------------------------------------------------------------
# H13 H5
# -------------------------------------------------------------------------------
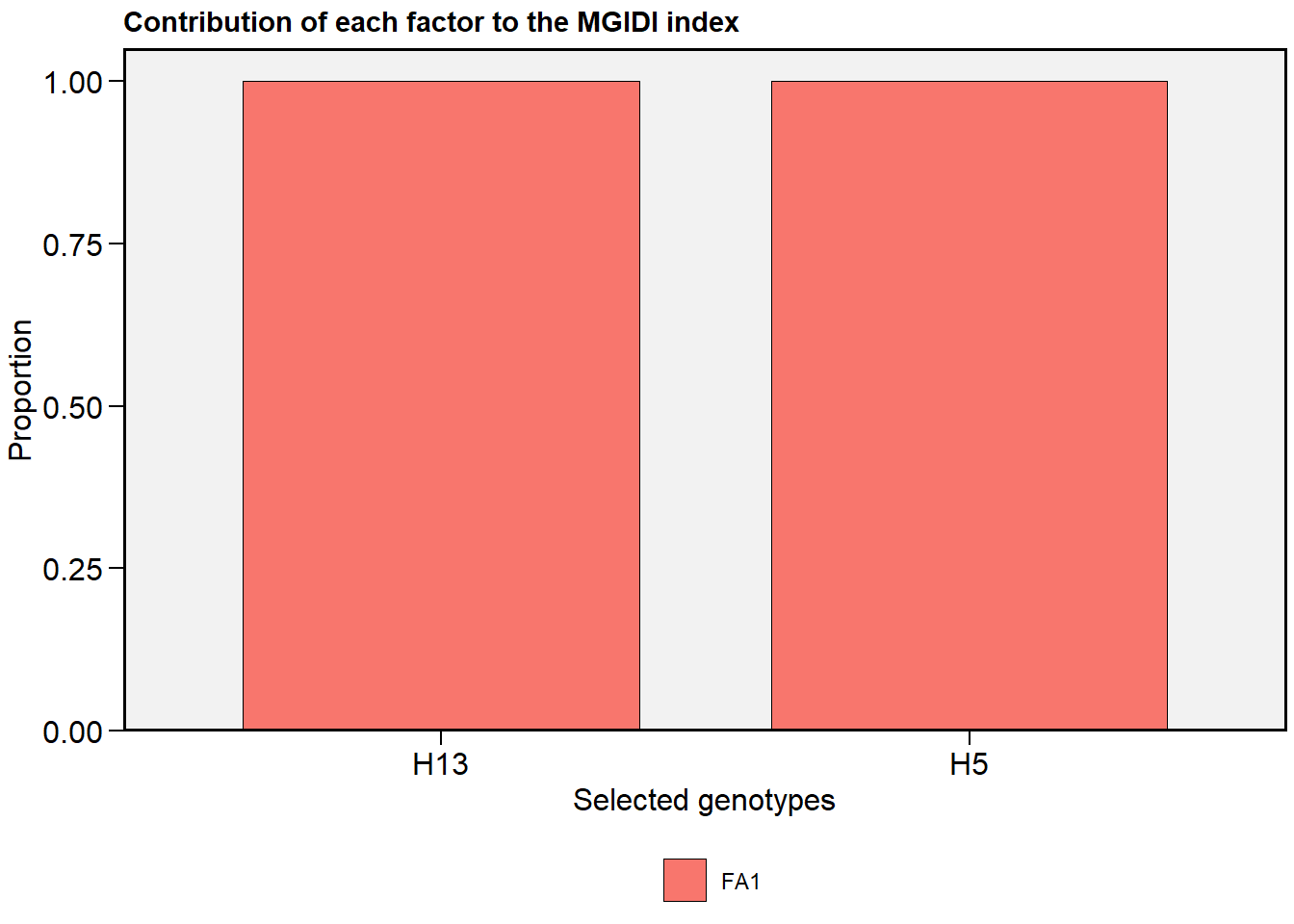
- The S3 method
plot()for objects of classmgidihas a new argumenttype = "contribution"to plot the contribution of each factor in the MGIDI index.
plot(mgidi_index, type = "contribution")
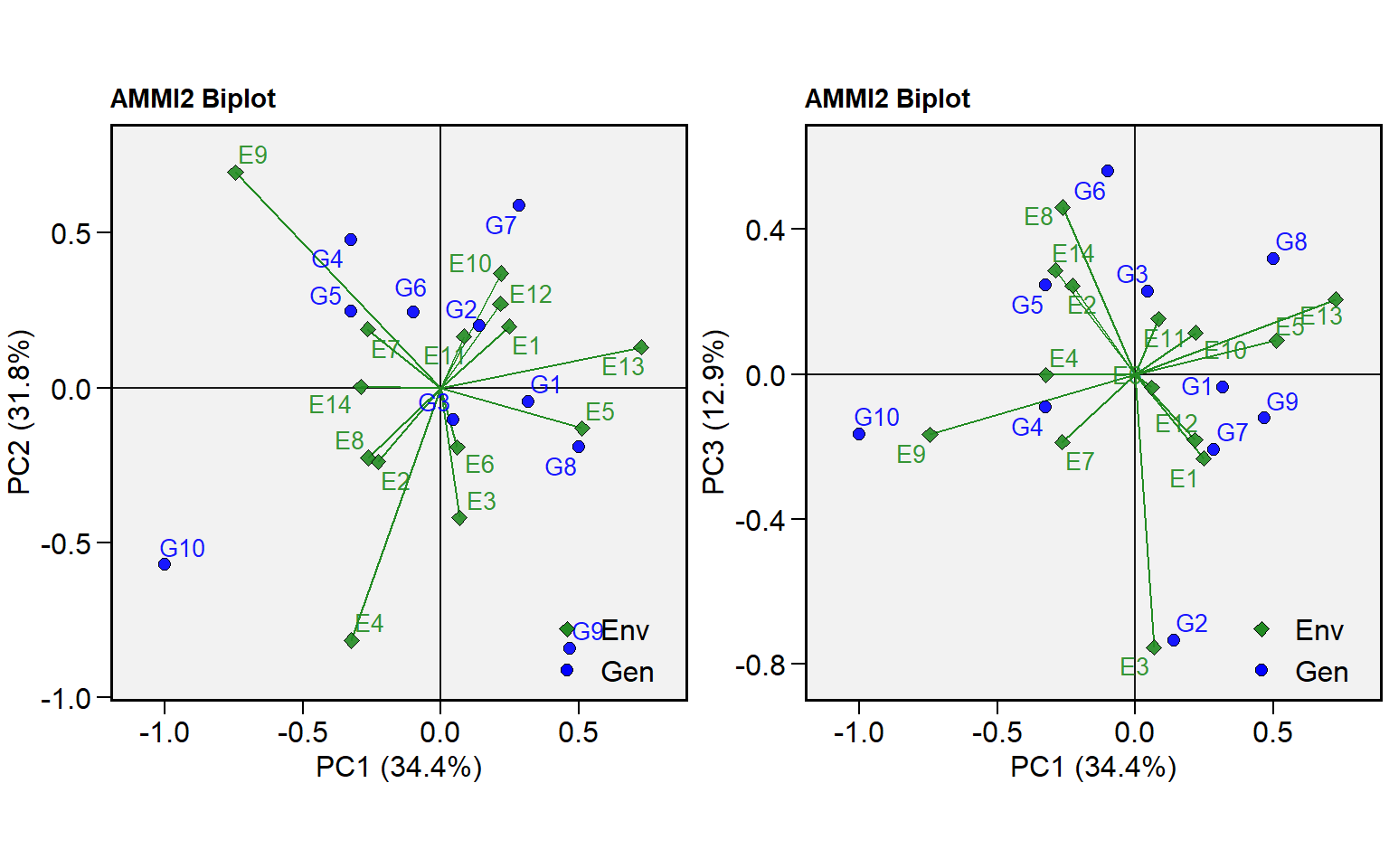
plot_scores()now can produce a biplot showing other axes besides PC1 and PC2. To change the default IPCA in each axis use the new argumentsfirstandsecond.
ammi <- performs_ammi(data_ge, ENV, GEN, REP, GY)
# variable GY
# ---------------------------------------------------------------------------
# AMMI analysis table
# ---------------------------------------------------------------------------
# Source Df Sum Sq Mean Sq F value Pr(>F) Proportion Accumulated
# ENV 13 279.574 21.5057 62.33 0.00e+00 . .
# REP(ENV) 28 9.662 0.3451 3.57 3.59e-08 . .
# GEN 9 12.995 1.4439 14.93 2.19e-19 . .
# GEN:ENV 117 31.220 0.2668 2.76 1.01e-11 . .
# PC1 21 10.749 0.5119 5.29 0.00e+00 34.4 34.4
# PC2 19 9.924 0.5223 5.40 0.00e+00 31.8 66.2
# PC3 17 4.039 0.2376 2.46 1.40e-03 12.9 79.2
# PC4 15 3.074 0.2049 2.12 9.60e-03 9.8 89
# PC5 13 1.446 0.1113 1.15 3.18e-01 4.6 93.6
# PC6 11 0.932 0.0848 0.88 5.61e-01 3 96.6
# PC7 9 0.567 0.0630 0.65 7.53e-01 1.8 98.4
# PC8 7 0.362 0.0518 0.54 8.04e-01 1.2 99.6
# PC9 5 0.126 0.0252 0.26 9.34e-01 0.4 100
# Residuals 252 24.367 0.0967 NA NA . .
# Total 536 389.036 0.7258 NA NA <NA> <NA>
# ---------------------------------------------------------------------------
#
# All variables with significant (p < 0.05) genotype-vs-environment interaction
# Done!
p1 <- plot_scores(ammi, type = 2)
p2 <- plot_scores(ammi, type = 2, second = "PC3")
arrange_ggplot(p1, p2)
Find out more about metan at https://tiagoolivoto.github.io/metan/