Análise GGE
O modelo de interação “genótipo + interação genótipo-ambiente (GGE)” tem sido amplamente utilizado para avaliação de genótipos e identificação de megaambientes em ensaios de múltiplos ambientes (MET). Este modelo considera um biplot GGE (ou seja, G + GE), que é construído pelos dois primeiros componentes principais em escala simétrica (PC1 e PC2) derivados da decomposição de valor singular de dados MET centrados no ambiente. O GGE biplot exibe graficamente G mais GE de um MET de uma forma que facilita a avaliação visual do genótipo e a identificação do megaambiente Yan (2007)1.
library(metan)
## Registered S3 method overwritten by 'GGally':
## method from
## +.gg ggplot2
## |=========================================================|
## | Multi-Environment Trial Analysis (metan) v1.15.0 |
## | Author: Tiago Olivoto |
## | Type 'citation('metan')' to know how to cite metan |
## | Type 'vignette('metan_start')' for a short tutorial |
## | Visit 'https://bit.ly/pkgmetan' for a complete tutorial |
## |=========================================================|
library(rio)
# gerar tabelas html
print_tbl <- function(table, digits = 3, ...){
knitr::kable(table, booktabs = TRUE, digits = digits, ...)
}
df_ge <- import("http://bit.ly/df_ge", setclass = "tbl")
O modelo GGE
O rendimento médio do genótipo * i * no ambiente * j * é comumente descrito por um modelo linear geral
$$ \hat y_{ij} + \mu + \alpha_i + \beta_j + \phi_ {ij} $$
onde \(\hat y_{ij}\) é o rendimento médio do genótipo \(i\) no ambiente \(j\), \(i = 1, … g; j = 1, … e \) sendo \(g\) e \(e\) os números de genótipos e ambientes, respectivamente; \(\mu \) é a média geral; \(\alpha_i \) é o efeito principal do genótipo \(i\); \(\beta_j \) é o efeito principal do ambiente \(j\), e \(\phi_{ij} \) é o efeito de interação entre o genótipo \(i\) e o ambiente \(j\). A decomposição por valores singulares de \(\phi_ {ij} \) resulta no modelo AMMI. A exclusão de \(\alpha_i \), permite que a variação explicada por este termo (do genótipo) seja absorvida pelo termo \(\phi_ {ij}\). No modelo GGE, o termo \(\alpha_i \) é excluído do modelo acima e, em seguida, a matriz de dados centrada no ambiente, \(\phi_ {ij} \), é submetida decomposição por valor singular2. Explicitamente, temos:
$$ {\phi_ {ij} = \hat y_ {ij}} - \mu - \beta_j = \sum \limits_ {k = 1} ^ p \xi_ {ik} ^ * \eta_ {jk} ^ * $$
onde \(\xi_ {ik} ^ * = \lambda_k ^ \alpha \xi_ {ik}\); \(\eta_ {jk} ^ * = \lambda_k ^ {1- \alpha} \eta_ {jk} \), sendo \(\lambda_k \) o \(k\)-ésimo autovalor (\(k = 1, … p\) , com \(p \le min (e, g)\); \(\alpha \) é o fator de partição de valor singular para o componente principal (PC) \(k\); \(\xi_{ik} ^ * \) e \(\eta_{jk} ^ * \) são os escores para o genótipo \(i\) e ambiente \(j\), respectivamente.
A função gge() é usada para ajustar um modelo GGE. De acordo com Yan (2003)2, a função suporta quatro métodos de centralização de dados, dois métodos de escalonamento de dados e três opções para particionamento de valor singular:
Métodos de centralização disponíveis
0ou"none"para nenhuma centralização;1ou"global"centrado globalmente (E + G + GE);2ou"environment"(padrão), para centrado no ambiente (G + GE);3ou"double"centrado na interação (GE), ou seja, o modelo AMMI. Um biplot não pode ser produzido com modelos produzidos sem centralização.
Métodos de escalonamento disponíveis
0ou"none"(padrão) para nenhum escalonamento;1ou"sd"onde cada valor é dividido pelo desvio padrão de seu ambiente correspondente (coluna). Isso colocará todos os ambientes aproximadamente na mesma faixa de valores.
Métodos de particionamento de valores singulares disponíveis
1ou"genotype"O valor singular é inteiramente particionado nos autovetores do genótipo, também chamados de preservação de métrica de linha;2ou"environment"(padrão) o valor singular é inteiramente particionado nos autovetores do ambiente, também chamados de preservação de métrica de coluna;3ou"symmetrical"O valor singular é simetricamente particionado nos autovetores de genótipo e do ambiente. Este SVP é mais frequentemente usado na análise AMMI e outras análises biplot, mas não é ideal para visualizar a relação entre as entradas ou que entre os testadores1.
Ajustando o modelo GGE
gge_model <-
gge(df_ge,
env = ENV,
gen = GEN,
resp = MMG)
O modelo acima foi ajustado considerando
- Preservação da métrica da coluna (onde o valor singular é inteiramente particionado nos autovetores do ambiente)
- Centrado no ambiente (o biplot conterá uma informação mista de G + GEI)
- Nenhum método de escalonamento.
Para alterar essas configurações padrão, use os argumentos svp, centering e scaling, respectivamente.
Visualizando o Biplot
A função genérica plot() é usada para gerar um biplot usando como entrada um modelo ajustado da classe gge. O tipo de biplot é escolhido pelo argumento type na função. Dez tipos de biplots estão disponíveis de acordo com @ Yan2003.
type = 1Um biplot básico.type = 2Desempenho médio vs. estabilidade.type = 3Qual-ganhou-onde.type = 4Discriminatividade vs. representatividade.type = 5Examinar um ambiente.type = 6Classificação de ambientes.type = 7Examinar um genótipo.type = 8Classificação de genótipostype = 9Comparar dois genótipos.type = 10Relação entre ambientes.
Neste material, para cada tipo de biplot, são produzidos dois gráficos. Um com as configurações padrão e outro para mostrar algumas opções gráficas da função.
Biplot tipo 1: Um biplot básico
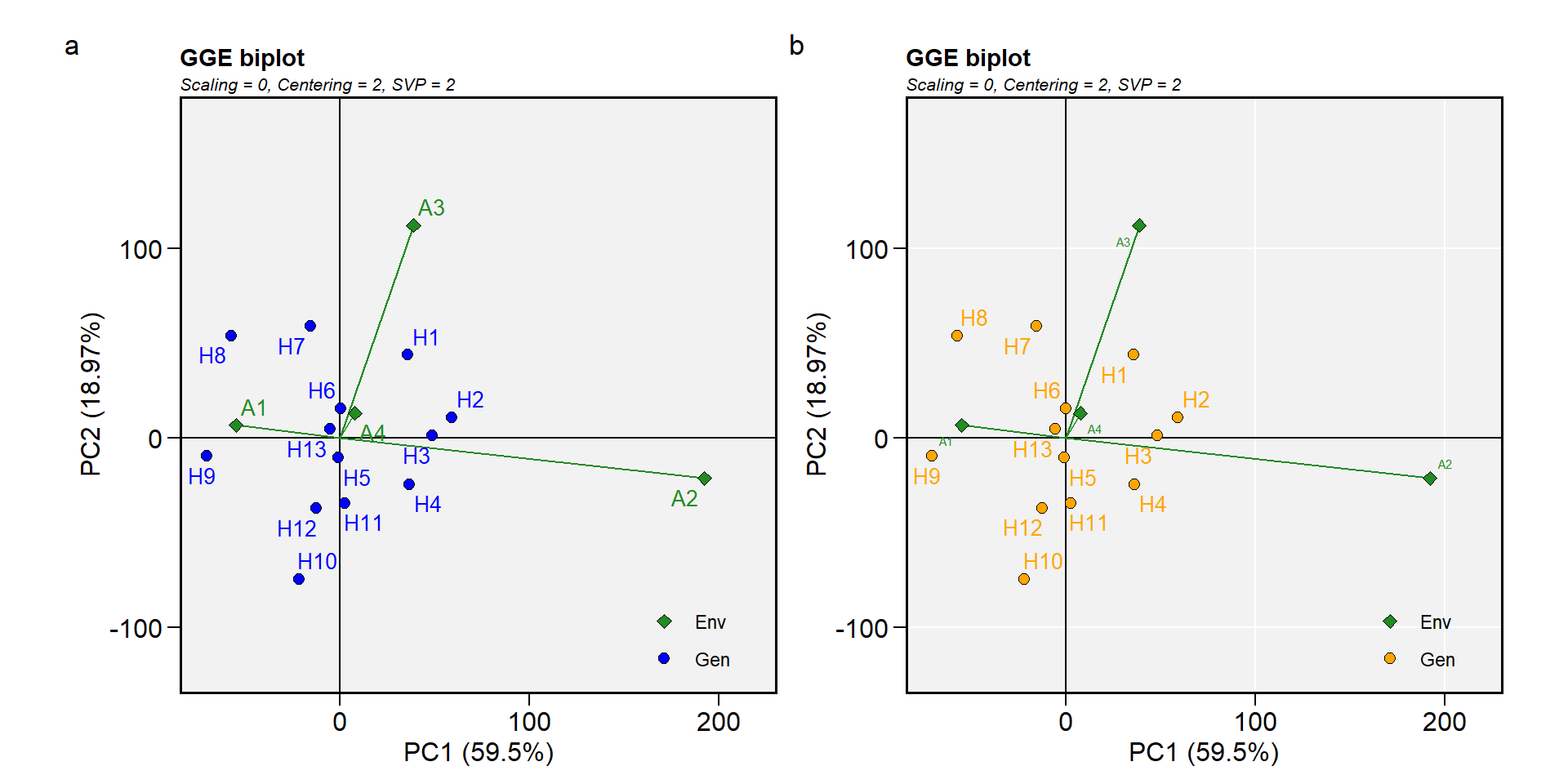
Esta é a configuração padrão no gráfico da função, portanto, este biplot é produzido apenas chamando plot(modelo), como mostrado abaixo.
a <- plot(gge_model)
b <- plot(gge_model,
col.gen = "orange",
size.text.env = 2,
plot_theme = theme_metan (grid = "both"))
arrange_ggplot(a, b, tag_levels = "a")
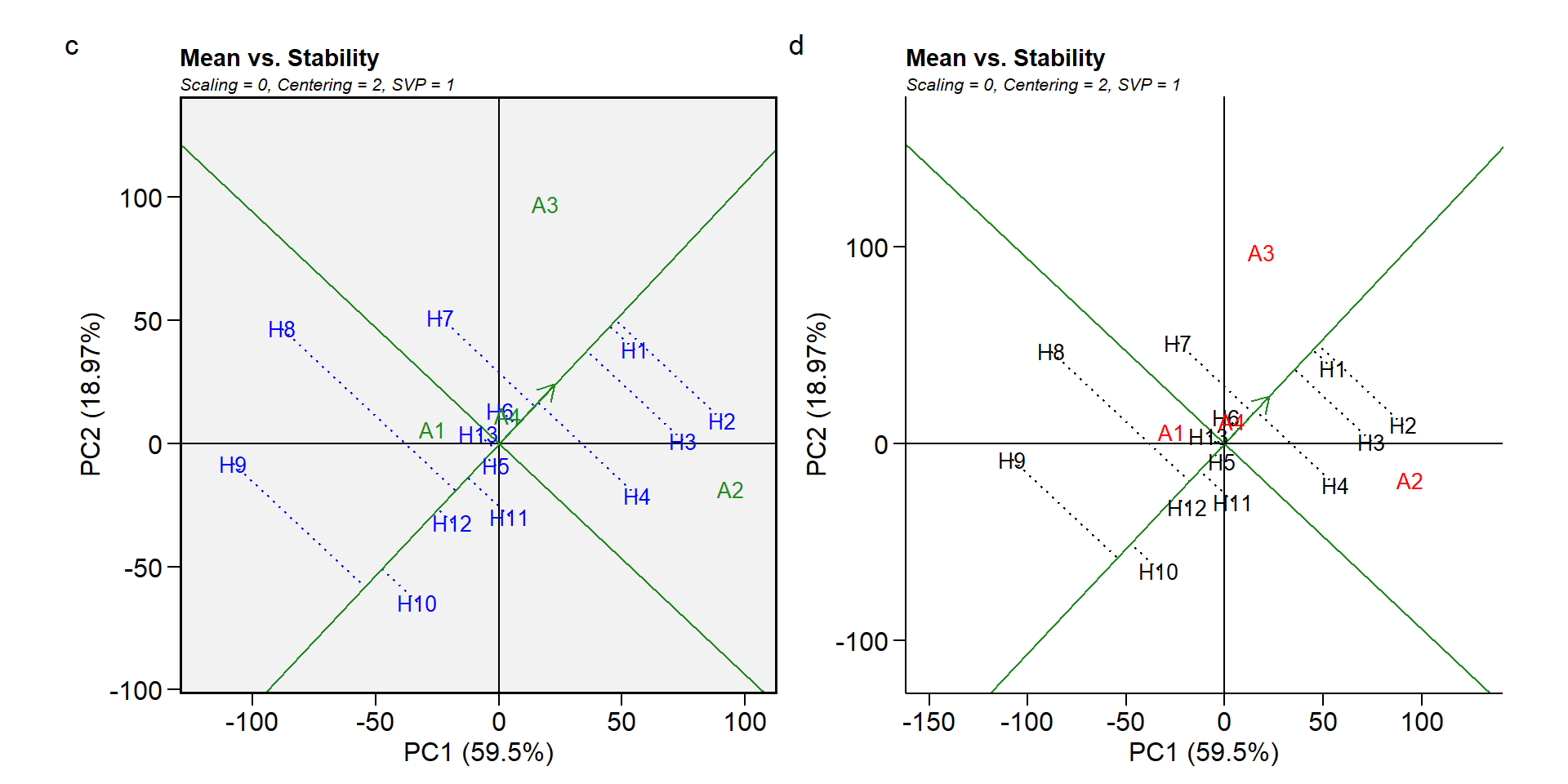
Biplot tipo 2: Desempenho médio vs. estabilidade
Neste biplot, a visualização da performance e da estabilidade dos genótipos é obtida desenhando uma coordenada ambiental média (AEC) no biplot focado no genótipo. Primeiro, um ambiente médio, representado pelo pequeno círculo, é definido pelos escores médios de PC1 e PC2 dos ambientes. A linha que passa pela origem do biplot e pelo AEC pode ser chamada de média. As projeções dos escores do genótipo neste eixo deve, portanto, aproximar o rendimento médio dos genótipos.
A ordenada AEC é a linha que passa pela origem do biplot e é perpendicular à abscissa AEC. Portanto, se a abscissa AEC representa o G, a ordenada AEC deve aproximar o GEI associado a cada genótipo, que é uma medida de variabilidade ou instabilidade dos genótipos. Uma maior projeção na ordenada AEC, independente da direção, significa maior instabilidade. Em nosso exemplo, H1 foi considerado relativamente estável e com boa performance.
gge_model <- gge (df_ge, ENV, GEN, MMG, svp = "genotype")
c <- plot(gge_model, type = 2)
d <- plot(gge_model,
type = 2,
col.gen = "black",
col.env = "red",
axis_expand = 1.5,
plot_theme = theme_metan_minimal())
arrange_ggplot(c, d, tag_levels = list(c("c", "d")))
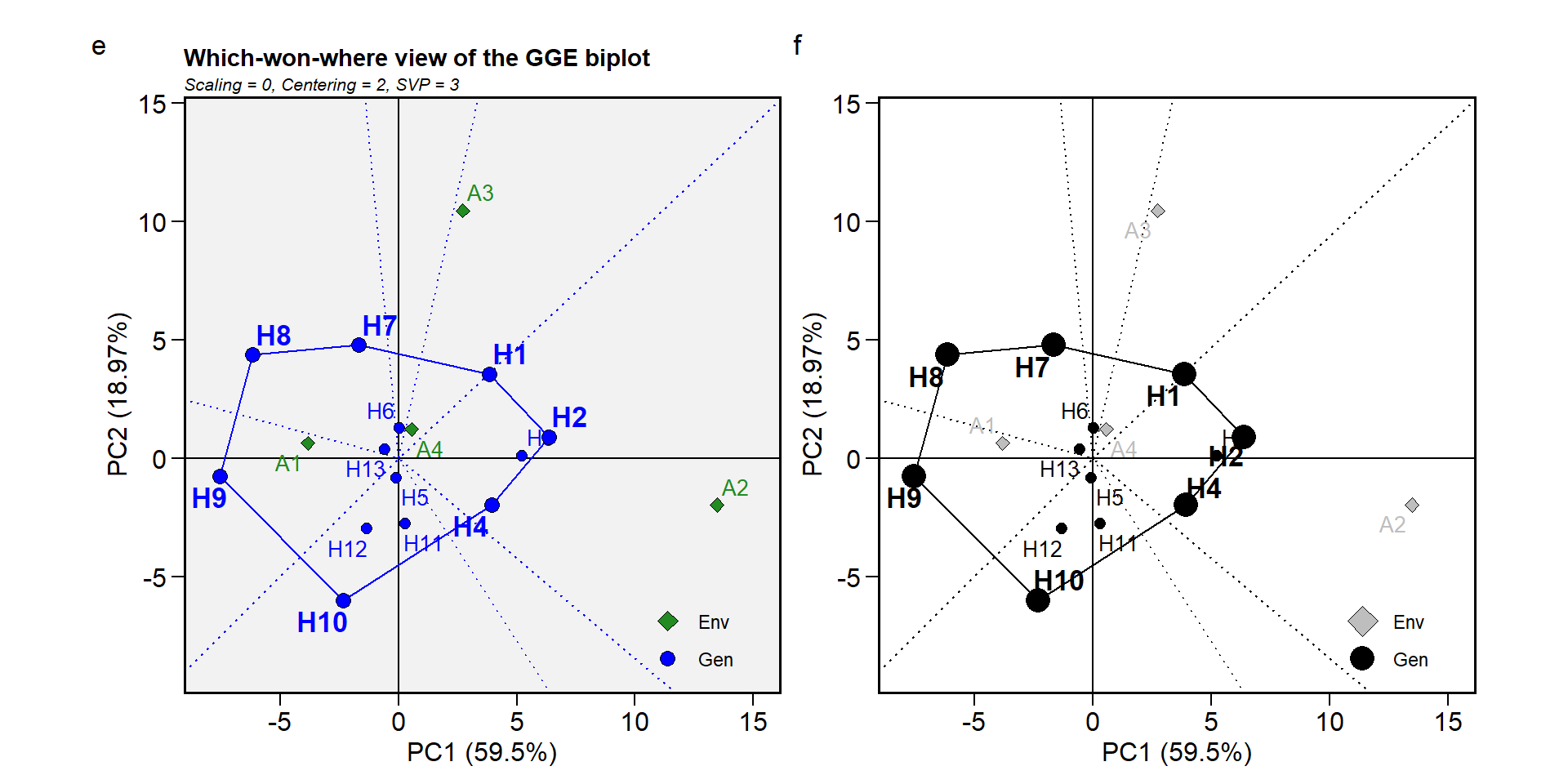
Biplot tipo 3: Qual-Ganhou-Onde
Neste biplot, um polígono é desenhado unindo os genótipos que estão localizados mais longe da origem do biplot, de modo que todos os outros genótipos estão contidos no polígono. Os genótipos-vértice possuem os vetores mais longos, em suas respectivas direções, o que é uma medida de capacidade de resposta aos ambientes. Eles estão, portanto, entre os genótipos mais responsivos; todos os outros genótipos são menos responsivos em suas respectivas direções. Um genótipo localizado na origem teria a mesma classificação em todos os ambientes.
As linhas perpendiculares às laterais do polígono dividem o biplot em setores. Como regra, o genótipo-vértice é o genótipo de maior rendimento em todos os ambientes que compartilham o setor com ele1.
gge_model <- gge (df_ge, ENV, GEN, MMG, svp = "symmetrical")
e <- plot(gge_model, type = 3)
f <- plot(gge_model,
type = 3,
size.shape.win = 5,
large_label = 6,
col.gen = "black",
col.env = "gray",
title = FALSE,
plot_theme = theme_metan(color.background = transparent_color()))
arrange_ggplot(e, f, tag_levels = list (c("e", "f")))
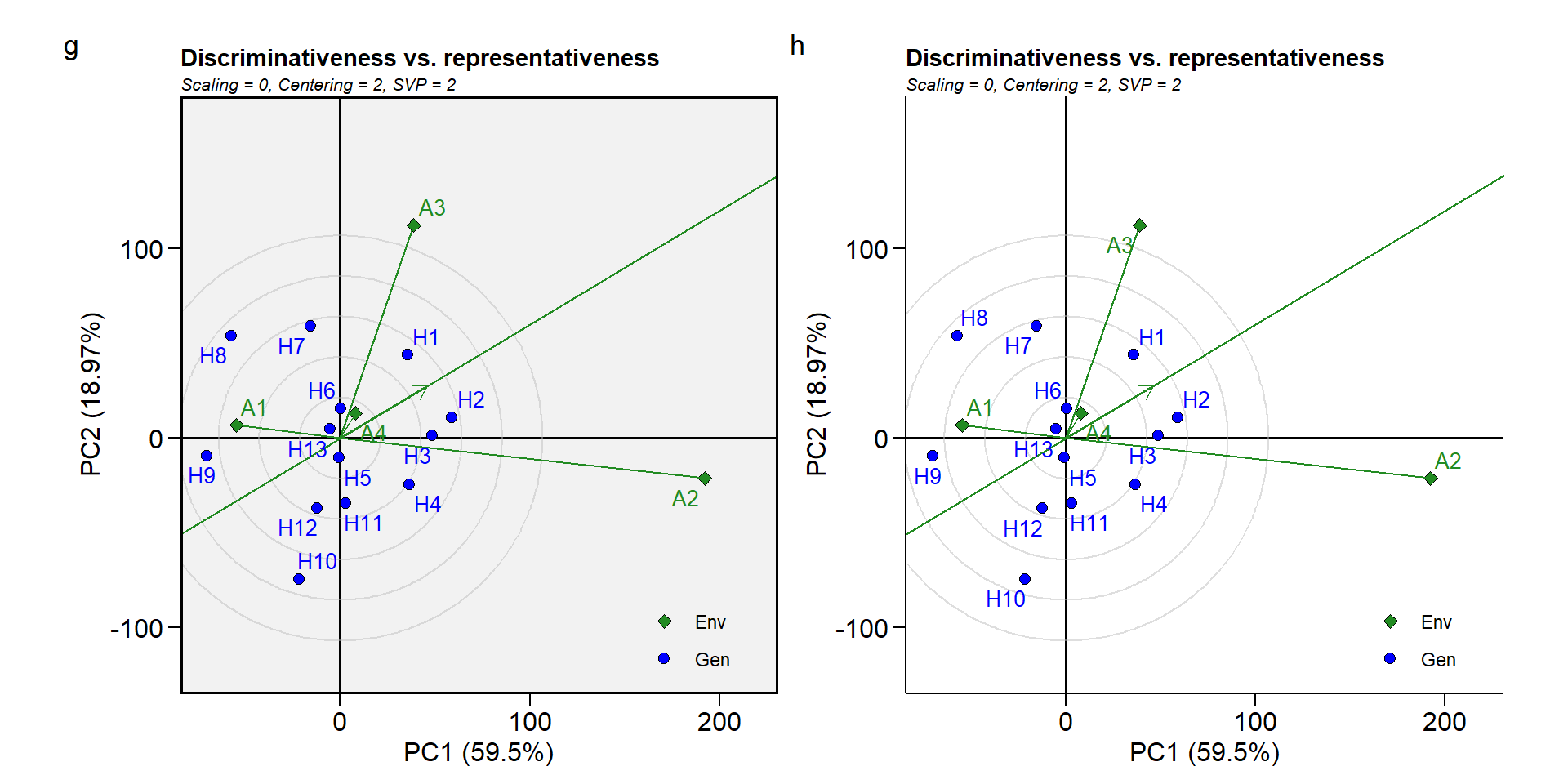
Biplot tipo 4: Discriminatividade vs. representatividade
A seta indica a posição do ambiente médio, que é definido pelas pontuações médias de PC1 e PC2 em todos os ambientes. Esse ambiente médio pode ser considerado um ambiente virtual. Uma linha que atravessa a origem do biplot e o ambiente médio, conhecido como eixo do ambiente médio. O comprimento do vetor de um ambiente representa sua capacidade de discriminação. O ângulo entre um ambiente e o eixo do ambiente médio representa a representatividade do ambiente: quanto maior o ângulo, menos representativo é o ambiente3
gge_model <- gge (df_ge, ENV, GEN, MMG)
g <- plot(gge_model, type = 4)
h <- plot(gge_model,
type = 4,
plot_theme = theme_metan_minimal())
arrange_ggplot(g, h, tag_levels = list (c ("g", "h")))
Biplot tipo 5: Examinar um ambiente
A identificação de genótipos mais adaptados a um ambiente pode ser facilmente alcançada por meio de um biplot GGE. Por exemplo, para visualizar o desempenho de diferentes genótipos em um determinado ambiente, por exemplo, A2, basta desenhar uma linha que passa pela origem do biplot e o marcador de A2. Os genótipos podem ser classificados de acordo com suas projeções no eixo A2 com base em seu desempenho em A2, na direção apontada pela seta. Em nosso exemplo, em A2, o genótipo de maior rendimento foi o H2, e o genótipo de menor rendimento foi o H9.
gge_model <- gge (df_ge, ENV, GEN, MMG, svp = "symmetrical")
i <- plot(gge_model, type = 5, sel_env = "A2")
j <- plot(gge_model,
type = 5,
sel_env = "A2",
col.gen = "black",
col.env = "black",
size.text.env = 12,
axis_expand = 1.5)
arrange_ggplot(i, j, tag_levels = list(c("i", "j")))
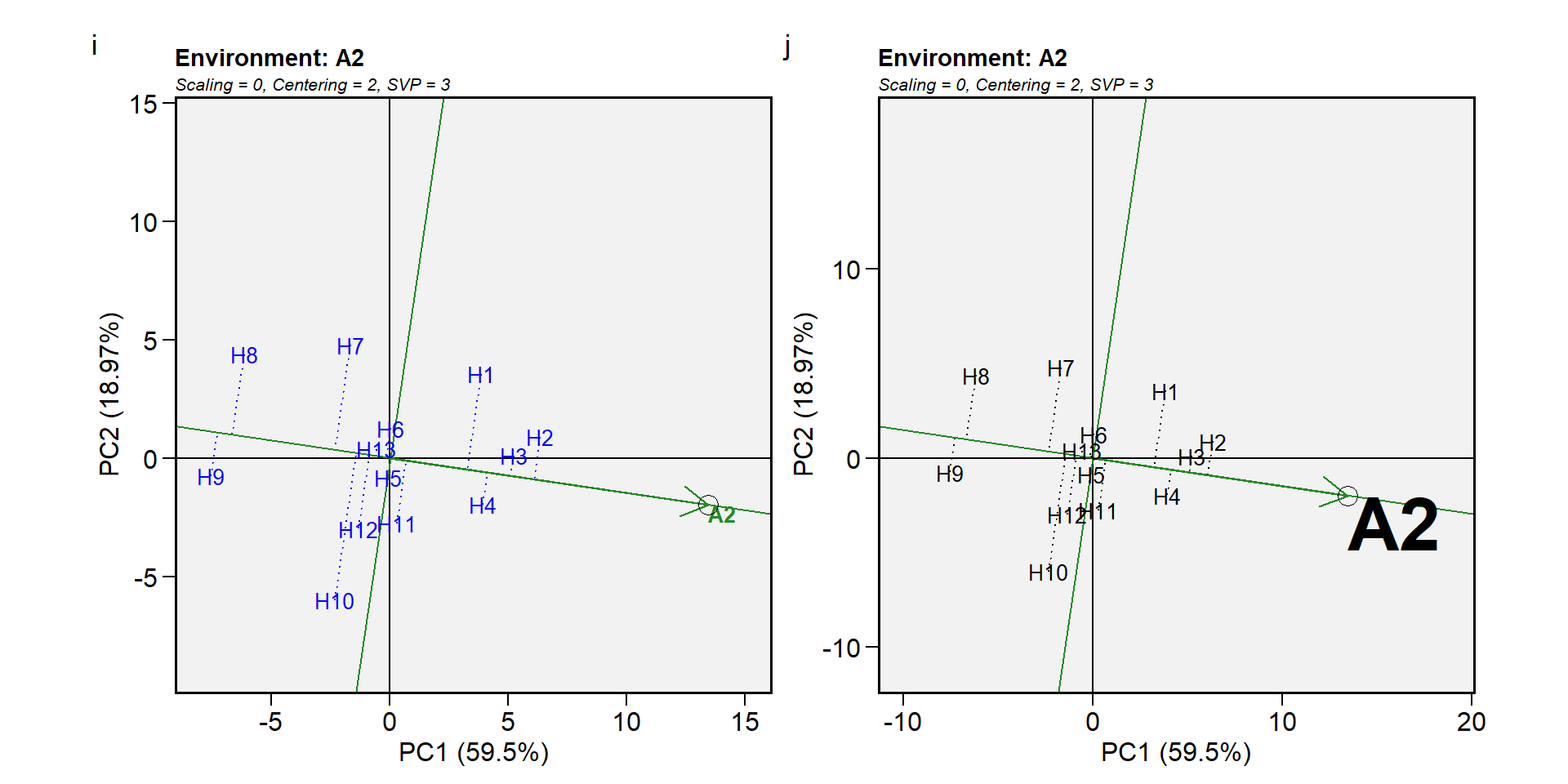
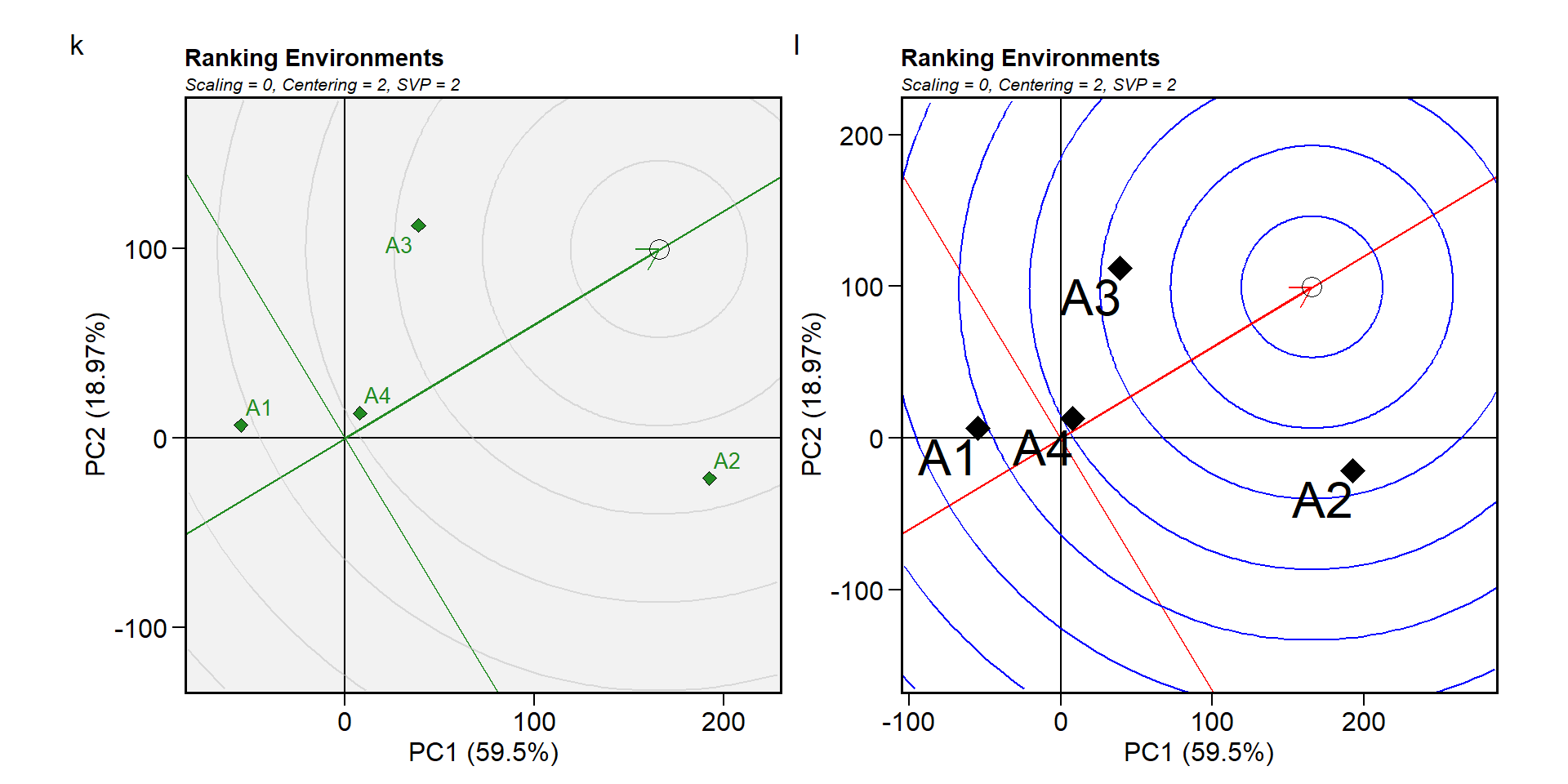
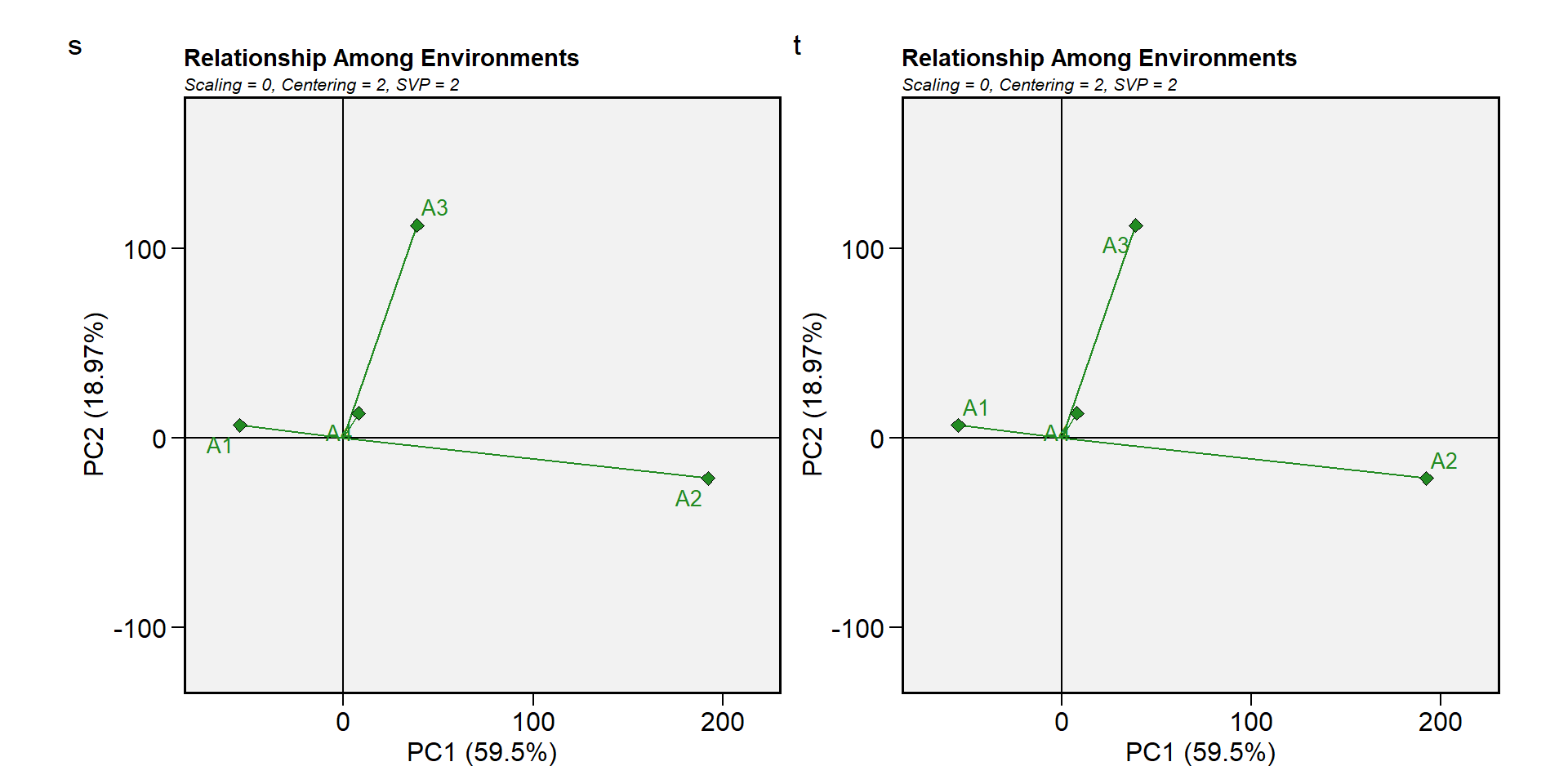
Biplot tipo 6: Ranqueamento de ambientes
Neste biplot, o ambiente “ideal” é usado como o centro de um conjunto de linhas concêntricas que servem como régua para medir a distância entre um ambiente e o ambiente ideal. Uma vez que o foco principal neste biplot são os ambientes, a partição de valor singular usada é “ambiente” (padrão).
gge_model <- gge(df_ge, ENV, GEN, MMG)
k <- plot(gge_model, type = 6)
l <- plot(gge_model,
type = 6,
col.line = "red",
col.gen = "black",
col.env = "black",
col.circle = "blue",
col.alpha.circle = 1,
size.text.env = 8,
axis_expand = 1.5,
size.shape = 4,
plot_theme = theme_metan(color.background = "white"))
arrange_ggplot(k, l, tag_levels = list (c ("k", "l")))
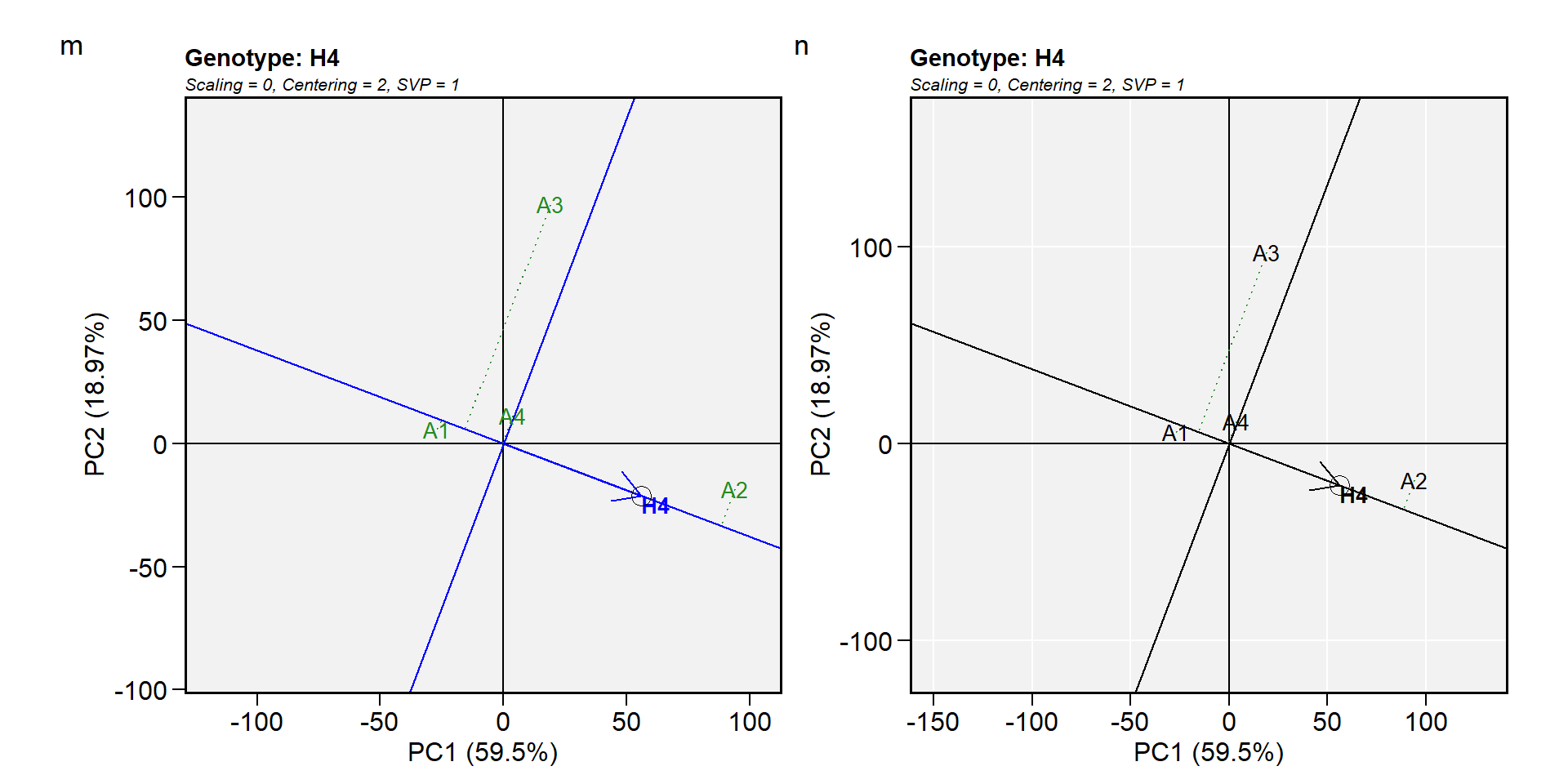
Biplot tipo 7: Examinar um genótipo
De forma análoga à visualização do desempenho do genótipo em um determinado ambiente (biplot 5), a visualização da média e da estabilidade dos genótipos é obtida desenhando uma coordenada de ambiente média (AEC) no biplot focado no genótipo.
gge_model <- gge(df_ge, ENV, GEN, MMG, svp = "genotype")
m <- plot(gge_model, type = 7, sel_gen = "H4")
n <- plot(gge_model,
type = 7,
sel_gen = "H4",
col.gen = "black",
col.env = "black",
size.text.env = 10,
axis_expand = 1.5,
plot_theme = theme_metan (grid = "both"))
arrange_ggplot(m, n, tag_levels = list(c("m", "n")))
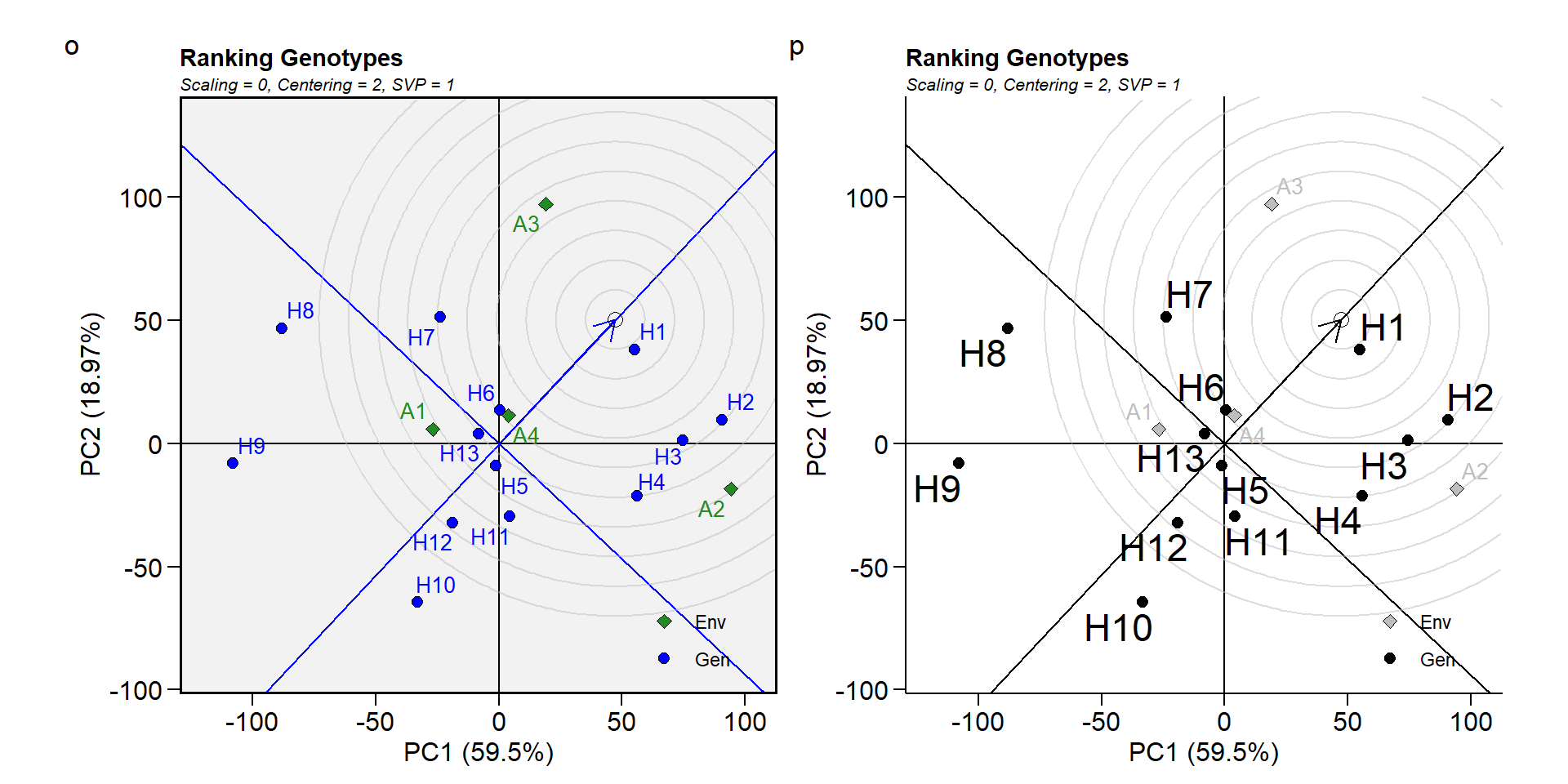
Biplot tipo 8: Classificação de genótipos
Este biplot compara todos os genótipos com o genótipo “ideal”. O genótipo ideal, representado pelo círculo pequeno com uma seta apontando para ele, é definido como tendo o maior rendimento em todos os ambientes. Ou seja, tem o rendimento médio mais alto e é absolutamente estável. Os genótipos são classificados com base em sua distância do genótipo ideal1. Em nosso exemplo, H1 superaram os outros genótipos.
o <- plot(gge_model, type = 8)
p <- plot(gge_model,
type = 8,
col.gen = "black",
col.env = "gray",
size.text.gen = 6,
plot_theme = theme_metan_minimal())
arrange_ggplot(o, p, tag_levels = list (c ("o", "p")))
Biplot tipo 9: Comparar dois genótipos
Para comparar dois genótipos, por exemplo, H10 e H8, desenhe uma linha para conectá-los e também uma perpendicular que passa pela origem do biplot e é perpendicular à linha que conecta esses genótipos. Vemos que um ambiente, A2, está do mesmo lado da linha perpendicular que H10, e os outros 03 ambientes estão do outro lado da linha perpendicular, junto com H8. Isso indica que H10 rendeu mais do que H8 em A2, mas H8 rendeu mais do que H10 nos outros 03 ambientes.
gge_model <- gge (df_ge, ENV, GEN, MMG, svp = "symmetrical")
q <- plot(gge_model,
type = 9,
sel_gen1 = "H8",
sel_gen2 = "H10")
r <- plot(gge_model,
type = 9,
sel_gen1 = "H8",
sel_gen2 = "H10",
col.gen = "black",
size.text.gen = 1,
size.text.win = 5,
title = FALSE,
plot_theme = theme_metan_minimal())
arrange_ggplot(q, r, tag_levels = list (c("q", "r")))
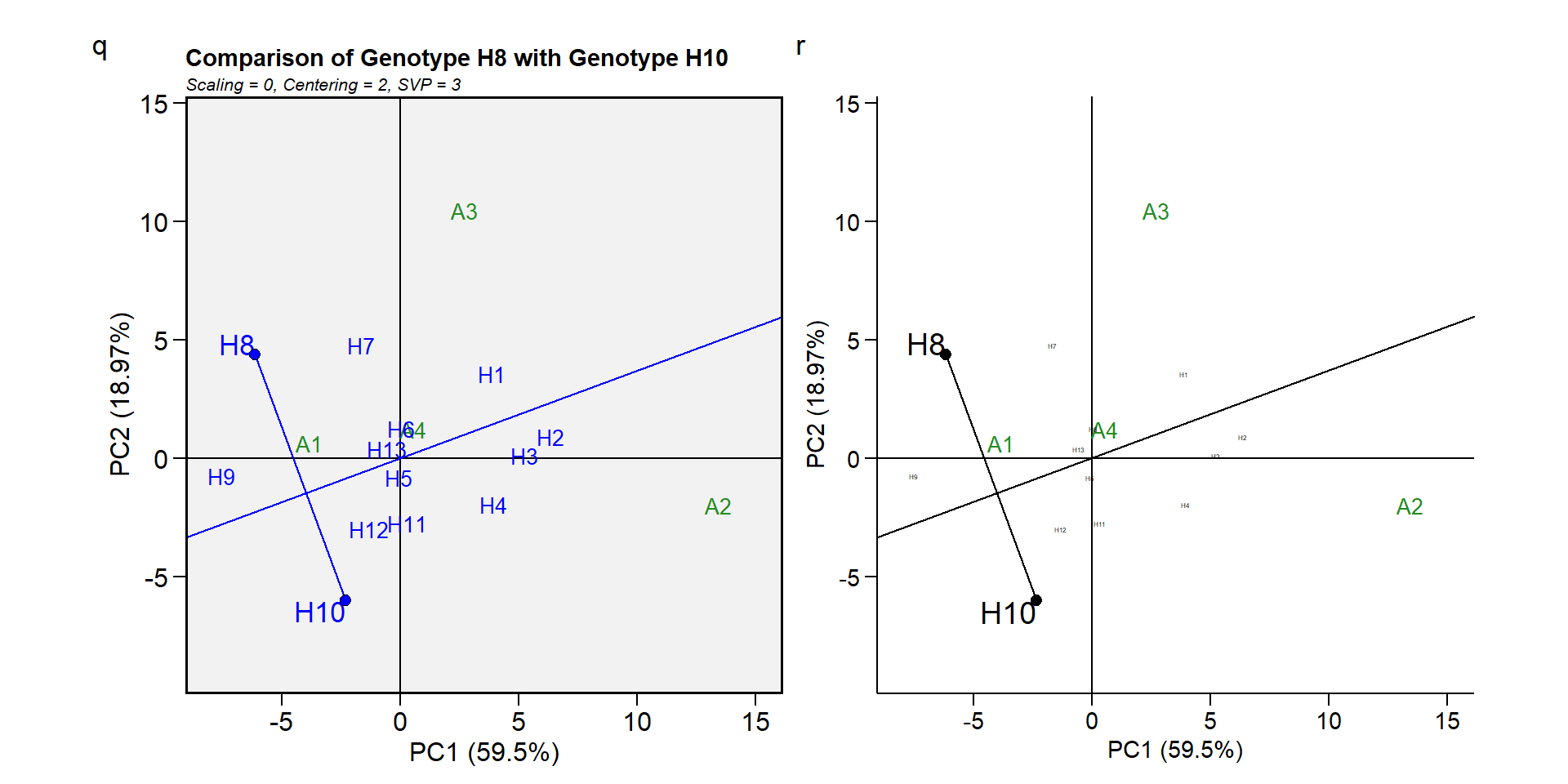
Biplot tipo 10: Relação entre ambientes
gge_model <- gge(df_ge, ENV, GEN, MMG)
s <- plot(gge_model, type = 10)
t <- plot(gge_model,
type = 10,
col.gen = "black",
título = FALSE)
arrange_ggplot(s, t, tag_levels = list (c("s", "t")))
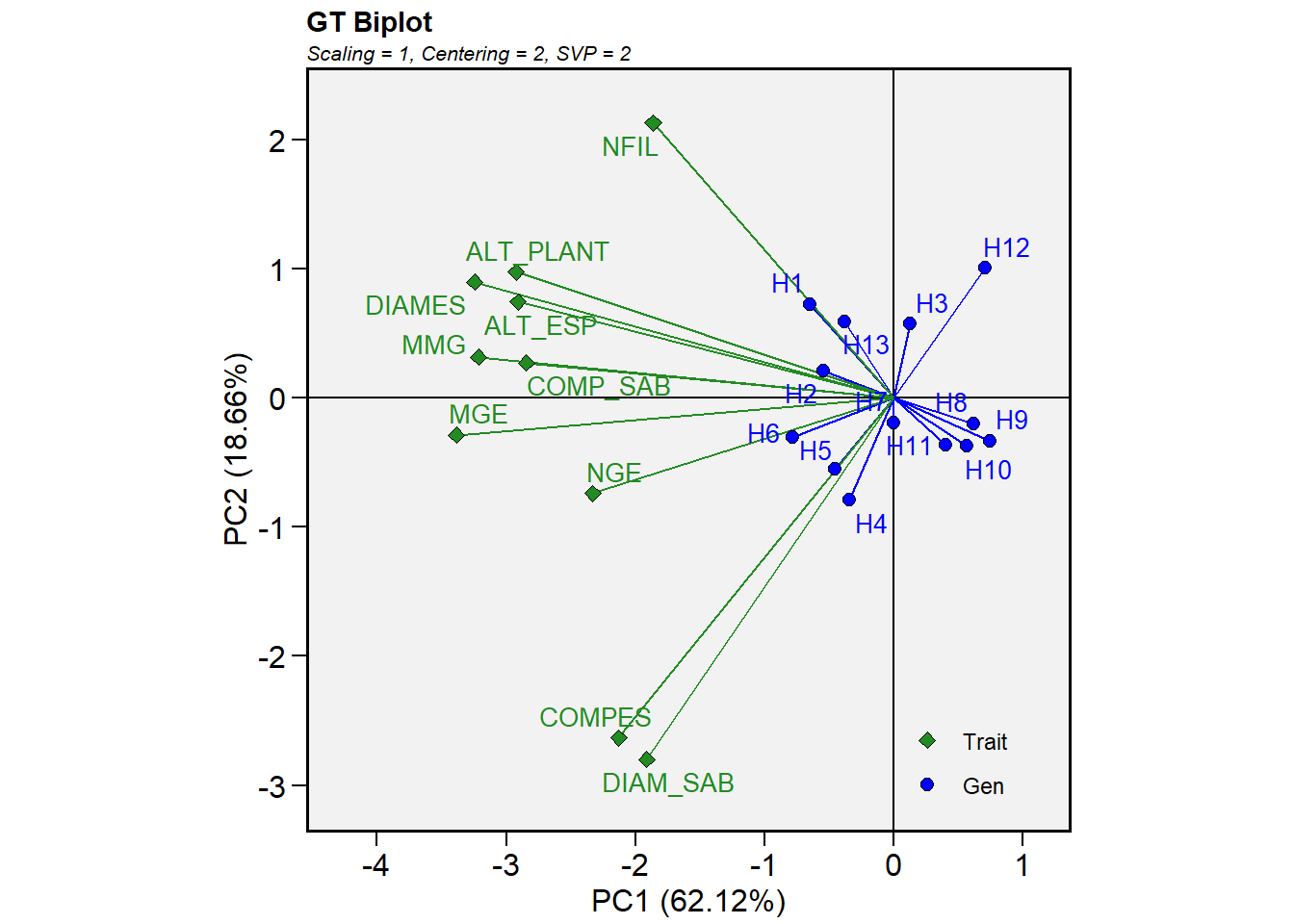
Genotype by trait biplot
# GT biplot for all numeric variables
mod <- gtb(df_ge, GEN, resp = everything())
plot(mod)
-
Yan, W., Kang, M. S., Ma, B., Woods, S., & Cornelius, P. L. (2007). GGE Biplot vs. AMMI analysis of genotype-by-environment data. Crop Science, 47(2), 641–653. https://doi.org/10.2135/cropsci2006.06.0374 ↩︎
-
Yan, Weikai., & Kang, M. S. (2003). GGE biplot analysis: A graphical tool for breeders, geneticists, and agronomists. CRC Press. ↩︎